Molecular Dynamics Simulation of Polymers by Lammps
Other than Gromacs, Lammps is another popular open-sourced package for molecular dynamics simulations. Here using coarse grained (CG) polymers as an example, the MD simulations of bottlebrush polymers (BBPs) is demonstrated.
Basic steps and input files
The basic steps to conduct MD simulations in Lammps is similar to that in Gromacs. The input files needed for a MD simulation in Lammps include:
- Data file containing the system information
- Input file for evolving molecular dynamics of the system
- A separated parameter files for the bonded and nonbonded parameters (can also be included in the Input file)
The Bead-spring model
The bead-spring model is a classical coarse-grained polymer model (a CG bead for each repeat unit connected by a spring). It has two parts: a repulsive WCA potential for pair interaction, namely shifted and truncated LJ potential, and the finite-extensible non-linear (FENE) potential for the bond potential (spring) to model the maximum stretch feature of polymers. Specifically, it has the following form:
where ε and σ are the potential well and distance, rc=21/6σ is the cutoff distance, beyond which the potential vanishes.
The FENE potential reads:
where k=30 ε/σ2 is the bond stiffness and r0=1.5σ is the maximum bond length.
The system is evolved by the Langevin dynamics:
where γ is the damping constant and R(t) is a delta-correlated stationary Gaussian process, satisfying:
The equilibration of bottlebrush polymer
Here, a BBP is equilibrated by Langevin dynamics using the potential described above. The input script can be found in many tutorials, for example ref [1,2]. The equilibrated structure along with its initial configuration of a bottlebrush polymer (backbone polymerization=100, side chain polymerization=8 with a grafting density of 1 per backbone bead) is:
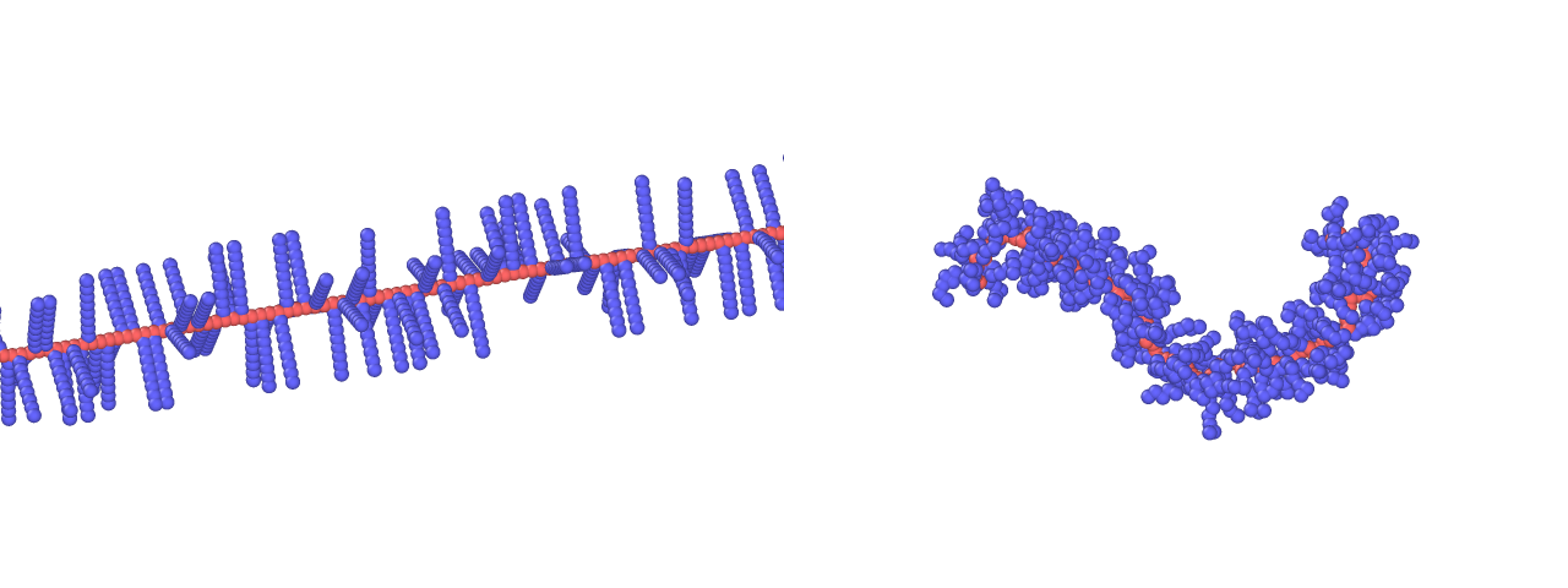
The animation is rendered as follows using ovito:

References
- Lammps tutorial of polymer [link].
- Another tutorial.