MD Simulations in Gromacs
With the coordinate and topology files prepared (last tutorial), molecular dynamics simulations can be carried out in Gromacs. The input files needed for a MD simulation in Gromacs include:
- Coordinate file containing the PEO polymer and solvent (SPCE water)
- Topology file, namely, topol.top file and the other itp files included therein
- MD parameter file controlling the simulations (integration, temperature, pressure control, etc.)
Simulation Protocol
Depending on the complexity and environment where the system is in, the protocol can be very different. Generally, there are two principal steps: equilibration and production run. Some systems may be complex and need multi-steps equilibration. In this example, a four-step protocol is employed for the PEO in water simulation:
- Energy minimization of the solvated PEO-water system
- nvt equilibration (canonical ensemble)
- npt equilibration (isothermal-isobaric ensemble)
- Production run in isothermal-isobaric ensemble
The concrete Gromacs (version 2021.4) commands to carry out the aforementioned simulation can be summarized as:
#!/bin/bash
# create the system
gmx editconf -f PEO4.gro -box 2.5 2.5 2.5 -o PEO4_newbox.gro
# solvate
gmx solvate -cp PEO4_newbox.gro -cs spc216.gro -p topol.top -o PEO4_solv.gro
# minimize
gmx grompp -f minim.mdp -c PEO4_solv.gro -p topol.top -o em.tpr
gmx mdrun -deffnm em -v
# nvt
gmx grompp -f nvt.mdp -c em.gro -p topol.top -o nvt.tpr
gmx mdrun -deffnm nvt -v
# npt
gmx grompp -f npt.mdp -c nvt.gro -p topol.top -o npt.tpr -t nvt.cpt
gmx mdrun -deffnm npt -v
# production
gmx grompp -f md.mdp -c npt.gro -p topol.top -o md.tpr -t npt.cpt
gmx mdrun -deffnm md -v
The parameter files (mdp files) can be found at Justin A. Lemkul’s website with minor modifications for own simulations. Beginners are strongly recommended to the Gromacs tutorials by him (link in the bottom line).
The movie of a PEO4 chain in water in 1ns production run is rendered as follows using vmd:
Data Analysis
After MD simulations, one can conduct post-analysis of quantities of interest. There are a bunch of handy commands for this end in Gromacs. For example, gmx energy for energy monitor, gmx density for density monitor, etc.
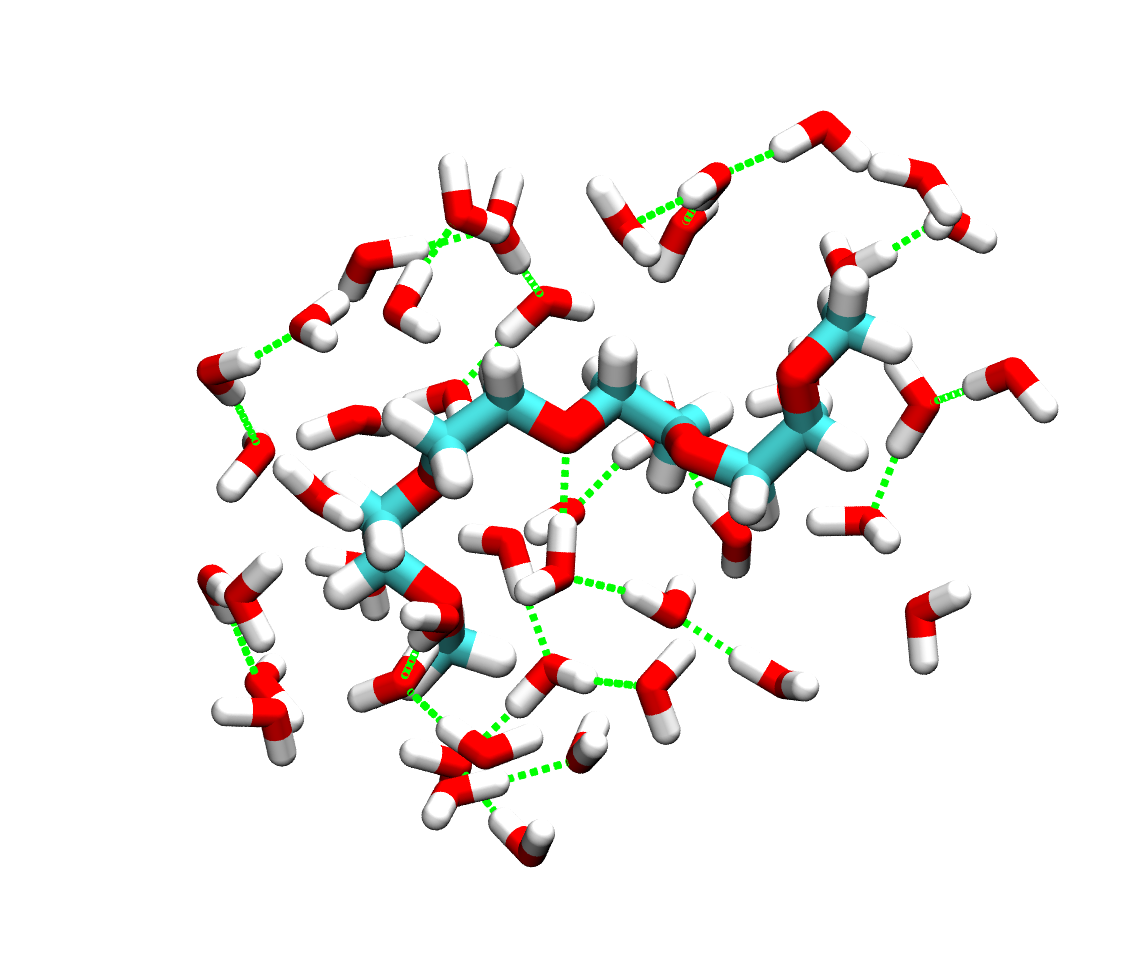
For simplicity, the hydrogen-bonding within the hydration shell of PEO is displayed:
Useful reading
The general background information about this tutorial can be found, for example, at: